

Kevin Guo, Rebecca Hawkins, and Bonnie Wu
Great Valley High School, 225 N. Phoenixville Pike, Malvern, PA 19355
Janssen Research and Development, 1400 McKean Road, PO Box 776, Spring House, PA 19477
Address correspondence to: kguo20@student.gvsd.org
ABSTRACT
Gold nanoparticles (GNPs) can create heat and free radicals after absorbing high energy photons from radiation. This light absorptive property has been utilized in cancer radiotherapy to produce highly localized heating and destroy cancer cells. For specific tumor accumulation, GNPs can be attached to monoclonal antibodies (mAbs) that recognize cell surface proteins overexpressed in certain types of cancer. One of these surface receptors is HER2, which is overexpressed in 20 - 25% of breast cancers and, therefore, constituted a suitable target for such mAb-driven GNP tumor localization. However, many mAbs cannot cross the cell membrane efficiently. To enable the efficient shuttling of GNPs into cancer cells, protein engineering technology was implemented to fuse penetratin, a cell-penetrating peptide (CPP), to the C-terminus of a wide-type anti-HER2 antibody heavy chain, generating a cell-penetrating mAb specific to the HER2 receptor. One of the recombinant mAbs (mAb-2CPP) completely preserved the HER2-binding activity and could promote the internalization of the antibody into MCF-7 and PC3 cancer cells that express HER2 on the cell surface. Both cell lines were then treated with GNPs conjugated to wild-type mAb (mAb-GNP) or mAb-CPP fusion (mAb-2CPP-GNP). The MAb-2CPP-GNP complex generated a higher mortality rate than mAb-GNP, likely due to more accumulation of GNPs in the cells. The study also investigated the interaction between X-ray radiation and GNPs, demonstrating that GNPs augmented the efficacy of X-ray radiation. Thus, a much lower dose of radiation could be used to generate a similar cell mortality rate. More GNPs were carried by mAb-2CPP into cancer cells and increased the cell mortality rate in a dose-dependent manner. Thus, an effective cell-penetrating antibody could promote the delivery of GNPs into target cancer cells and augment the efficacy of cancer radiotherapy with reduced side effects to patients.
INTRODUCTION
Cancer heat therapy, also referred to as nanoparticle hyperthermia, utilizes the strong light absorptive properties of gold nanoparticles (GNPs) to create heat and free radicals in a small localized region to burn away cancer after GNPs absorb high energy photons from radiation (Kaur et al., 2016). Therefore, GNPs have been considered as good candidates to enhance the effect of cancer radiation therapy (Jain et al., 2012; Hu et al., 2015; Saha et al., 2016).
Gold nanoparticles are gold coordination complexes with outstanding cytotoxic properties. When internalized into cells, these gold compounds can trigger direct mitochondrial damage and induce apoptosis (Gamberi, 2013). GNPs are small particles that can penetrate not only cancer cells, but also healthy tissues. Accumulation of these inherently toxic particles in healthy cells would lead to unwanted side effects and reduce the effective compound concentration at the tumor site. This lack of tumor selectivity can be addressed by attaching monoclonal antibodies (mAbs) that recognize specific cancer cell-surface proteins to the relatively large surface area of GNPs (Fay and Scott, 2011).
The HER2 receptor is overexpressed in about 30% of breast cancers and regulates important pathways involved in cell survival and proliferation. Therapeutic antibodies targeting the HER2 receptor have provided satisfactory results. Additionally, HER2-targeted nanoparticles were exploited to deliver drug to tumor sites (Mazzucchelli et al., 2014). Therefore, in this study, an anti-HER2 antibody targeting HER2 receptor was conjugated to GNPs in order to specifically deliver nanoparticles to breast cancer cells for photothermal ablation.
When conjugated to GNPs, mAbs will direct these nanoparticles to accumulate in target cancer cells. However, despite a highly specific and potent binder, mAbs cannot cross the cell membrane as efficiently as small compounds, thus limiting their use as targeting reagents to deliver GNPs into cancer cells.
Cell-penetrating peptides (CPPs) are a group of small peptides that can overcome the impermeability of the plasma membrane (Lim et al., 2013; Dinca et al., 2016; Ali et al., 2016). They are usually 10 to 30 amino acid (aa) peptides in length and are either arginine-rich and amphipathic, or lysine-rich and hydrophobic. CPPs can be linked to an antibody to promote its cellular uptake. When the cell translocation capacity of a CPP is paired with the targeting specificity of an antibody, the CPP-antibody complex is able to deliver the conjugated molecular cargo, i.e. drug compound, into target cells with high efficiency and specificity. In the previous project, penetratin, a 16-aa CPP with membrane translocation properties (Dupont et al., 2015; Watson et al., 2017), was conjugated to an anti-HER2 antibody using a chemical conjugation method via CLICK chemistry to promote the delivery of antibody-GNP complex into cancer cells. However, the chemical conjugation procedure was not only laborious with a poor yield, but also generated protein aggregation, which compromised the yield of the mAb-CPP complex (data not shown).
In this study, a protein engineering method was used to develop a cell-penetrating mAb specific to the breast cancer HER2 receptor by fusion of penetratin to either the C-terminus of the antibody heavy chain, or both the N-terminus and C-terminus of the heavy chain. While fusion to the N-terminus and C-terminus of heavy chain completely impaired the antibody binding, fusion of penetratin to the C-terminus of antibody heavy chain (hereafter referred as mAb-2CPP) completely preserved its HER2 binding activity and promoted the internalization of the anti-HER2 antibody into MCF-7 and PC3 cells expressing different levels of HER2 receptor on the cell surface. Furthermore, GNPs were conjugated to both the wild-type (mAb-GNP) and engineered mAb-2CPP fusion (mAb-2CPP-GNP) and were used to treat MCF-7 and PC3 cells. The cells were then exposed to X-ray radiation known for high energy photons and high penetration depth (Gianfaldoni et al., 2017). The MAb-2CPP-GNP complex consistently generated lower viability in both cell lines in a dose-dependent manner, indicating that mAb-2CPP fusion could promote more accumulation of GNPs in the cells. The results indicate that X-ray radiation could be used in combination with GNP to power cancer heat therapy. The presence of GNPs resulted in a similar cancer cell mortality rate even with a lower radiation dose. The study offers a novel approach to engineer cell-penetrating mAbs and expands their potential for therapeutic applications. More efficient delivery of GNPs into cancer cells would enhance the efficacy of radiation therapy and would allow the use of a lower treatment dose without compromising the efficacy, thereby reducing adverse side effects and risks to patients.
METHODS
Material and Reagents
MCF7 breast cancer cells overexpressing HER2, PC3 prostate cancer cells, and CHO cells were provided by Janssen Research and Development. DMEM cell culture media was purchased from ThermoFisher Scientific (Rockford, IL).
LinkOriented kit GOLD (Nanoimmunotech, Spain) is a chelate-modified gold nanoparticles bioconjugation kit designed for oriented immobilization of antibodies. The kit comprises 40 nm (average size) chelate-modified gold nanoparticles for metal-chelation to histidine-rich metal-binding sites present in the Fc domain of antibodies.
Recombinant anti-HER2 antibodies were cloned, expressed, and purified by Creative Biolabs (Shirley, NY) using an ExpiCHO-S cell expression system (ThermoFisher Scientific, Waltham, MA). The wild-type anti-HER2 antibody was provided by Creative Biolabs. The CellTiter-Glo® Luminescent Cell Viability Assay Kit was purchased from Promega (Madison, WI).
The reagents used for flow cytometry include biotin free Fc receptor blocker (Accurate Chemical, Westbury, NY), stain buffer (Becton Dickinson, Franklin Lakes, NJ), Phycoerythrin (PE) conjugated mouse anti-human IgG Fc (Southern Biotech, Birmingham, AL), and Cytofix™ Fixation Buffer (Becton Dickinson, Franklin Lakes, NJ).
For immunofluorescence and confocal imaging, cell tracker (ThermoFisher Scientific, Waltham, MA) was used to stain cells. Alexa fluor 488 conjugated wild-type and mAb-2CPP antibody were prepared by Creative Biolabs (Shirley, NY).
Equipment
The biological safety cabinet (Baker, Sanford, Maine) was used for cell culture. A centrifuge was employed during the conjugation of antibodies to GNPs. X-ray radiation was performed in a RS-2000 Pro Biological Irradiator (Rad Source, Buford, GA) at 19.53 Gray/min. An Envision plate reader (Perkin Elmer, Waltham, MA) was applied to capture the chemiluminescent signal generated from the CellTiter-Glo® cell viability assay. The Opera Phenix confocal microscope (Perkin Elmer, Waltham, MA) was implemented for cell imaging to monitor the entry of antibodies into cells. The binding of antibodies to MCF-7 or PC3 cells were characterized using flow cytometry (Becton Dickinson, Franklin Lakes, NJ). The plate shaker was procured from Lab-Line Instruments, Inc. (Melrose Park, IL).
Antibody-GNP Conjugation
Twenty micrograms of wild-type or recombinant anti-HER2 antibody were conjugated to 40 nm GNPs as per manufacturer guidelines. After the reagents were equilibrated to room temperature, an antibody solution was prepared at a final concentration of 60 μg/ml in the supplied reaction buffer. The antibody solution was then added to the microcentrifuge tube containing chelate-modified gold nanoparticles. The vials were agitated on a roller stirrer for 60 minutes at room temperature. The unbound antibody or protein was removed by centrifugation at 14,000 g for 10 minutes at 4oC. The supernatant was carefully removed without disturbing the beads and stored for later determination of the protein immobilization yield. The pellet of GNPs was resuspended in by 1 mL of reaction buffer and the wash steps were repeated three times. The GNPs immobilized with the antibody were resuspended in a suitable volume with reaction buffer. The conjugated antibody concentration was determined using the BCA protein assay.
Recombinant Protein Expression and Purification
Penetratin (16 aas, RQIKIWFQNRRMKWKK) was fused to the N-terminus and/or C-terminus of an anti-HER2 antibody heavy chain using molecular cloning technique. The wild-type and recombinant anti-HER2 antibody constructs were separately transfected to ExpiCHOS cell line for transient antibody expression. The ExpiCHOS cell line is a clonal derivative of the CHO-S cell line that has been selected for high protein expression. The expressed protein was secreted to the supernatant and purified through a protein A affinity column. The purified protein was stored in PBS buffer after buffer exchange via dialysis and concentration through a Centricon device.
MCF-7 and PC3 Cell Culture and Plating
MCF-7 and PC3 were cultured in DMEM in a T-75 flask at a 37°C CO2 incubator until 85% confluency was reached. For subculture, the flasks were removed from the incubator. After aspirating the media, cells in the flask were rinsed in 10 mL PBS followed by trypsin treatment for 3 minutes at room temperature. After dislodging, 7 mL of media were added to the flask to stop trypsin digestion. Cells were counted by viaCell, the Beckman automated cell counter, and then pelleted by centrifugation at 200 g for eight minutes. The cells were resuspended in a suitable volume of media and diluted to a defined concentration before they were seeded to the culture plates.
Flow Cytometry Analysis
MCF-7, PC3, and CHO cells (100,000 cells/well) were used for flow cytometry analysis to characterize the specific binding of antibodies to HER2 receptors on the cell surface. Cells were first incubated with blocking buffer in the dark for 30 minutes in a 96-well round bottom assay plate. After a wash in staining buffer, the cells were centrifuged at 400xg for five minutes followed by removal of supernatant. After resuspension in 50 µL of staining buffer with or without 0.5 µg of wild-type mAb, mAb-2CPP, or mAb-4CPP, the cells were stained for 30 minutes at 4°C. The cells were then washed in staining buffer and centrifuged at 400xg for 5 minutes. After removal of the supernatant, the cells were subsequently stained for 30 minutes at 4˚C in 100 µL of staining buffer containing 1 µL of Phycoerythrin (PE) conjugated mouse anti-human IgG Fc. After fixing with Cytofix™ Fixation Buffer and subsequent washing, the cells were analyzed using a flow cytometer.
Immunofluorescence and Confocal Imaging
The PC3 and CHO cells were seeded at 25,000 cells/well in a 96-well assay plate. The cells were immunostained using 1 µg/mL of Alexa fluor 488 conjugated wild-type or mAb-2CPP for 1 hour. After a wash, the media was replaced by fresh media containing cell tracker. After incubation for 20 minutes, the media was removed and replaced by fresh media for confocal imaging using Opera Phenix at 60X objective magnification using a 488 nm laser.
X-ray Radiation Treatment
X-ray radiation was performed by leaving cell culture plates in a RS-2000 Pro Biological Irradiator (Rad Source) at 19.53 Gray/min. A higher exposure time was used for a higher dose. For example, 10 Gray corresponds to 30 seconds of exposure time. The cell culture plates were returned to the 37 CO2 incubator before measurement of the cell viability.
CellTiter-Glo® Cell Viability Assay
The CellTiter-Glo® cell viability assay is a method used to determine the number of viable cells in culture based on quantitation of the level of cellular ATP, an indicator of metabolically active cells. The homogeneous “add-mix-measure” procedure lyses cells and generates a luminescent signal proportional to the amount of ATP present in the cells. In this study, 5 mL of CellTiterGlo® buffer were added into the bottle containing CellTiterGlo® substrate to make a 4X CellTiterGlo® assay solution. Subsequently 50µL CellTiterGlo® assay solution were dispensed into assay wells in a cell culture plate. After gently agitating on a plate shaker for two minutes, the plate was sealed with a transparent adhesive film and read in an Envision® plate reader to measure the luminescent signal.

Figure 1. Structure of engineered penetratin-antibody fusion proteins. The structure of the two recombinant mAb-CPP fusion proteins is illustrated in Figure 1A and Figure 1B. Figure 1A. Two copies of penetratin (16 amino acids) were fused on the C-terminus of the anti-HER2 mAb heavy chain. Thus, the mAb-2CPP carries two copies of penetratin sequence. Figure 1B. Two copies of penetratin were fused to the N-terminus and C-terminus of anti-HER2 mAb heavy chain, respectively. Hence, the mAb-4CPP carries four copies of penetratin sequence. The diagrams were adapted from the immunoglobulin structure.
RESULTS
Structure of Penetratin-mAb Fusion Proteins
Two versions of penetratin-mAb fusion proteins were generated via molecular cloning and recombinant protein expression (Fig. 1). For one penetratin-mAb fusion protein, mAb-2CPP, penetratin sequence was fused to the C-terminus of the anti-HER2 mAb heavy chain (Fig. 1A). As a result, the mAb-2CPP carried a total of two copies of penetratin sequence. For the other fusion protein, mAb-4CPP, penetratin was fused to both the N-terminus and the C-terminus of the anti-HER2 mAb heavy chain (Fig. 1B). Thus, the mAb-4CPP carried four copies of penetratin sequence. The incorporation of penetratin (CPP) reduced the expression level of recombinant protein in HEK293F. However, the protein expression level was substantially improved after the cell line was switched to ExpiCHOS, an optimized CHO cell line that delivers high-titer protein production (Zhong et al., 2018). A higher than expected level of protein loss was observed during buffer exchange and the subsequent protein concentration. It seemed that the recombinant antibodies had a tendency to stick to the membrane surface of the dialysis tube and concentration column. Further optimization of these steps by optimizing the devices used for dialysis and concentration would improve the protein recovery and maximize the yield of purified recombinant antibodies.
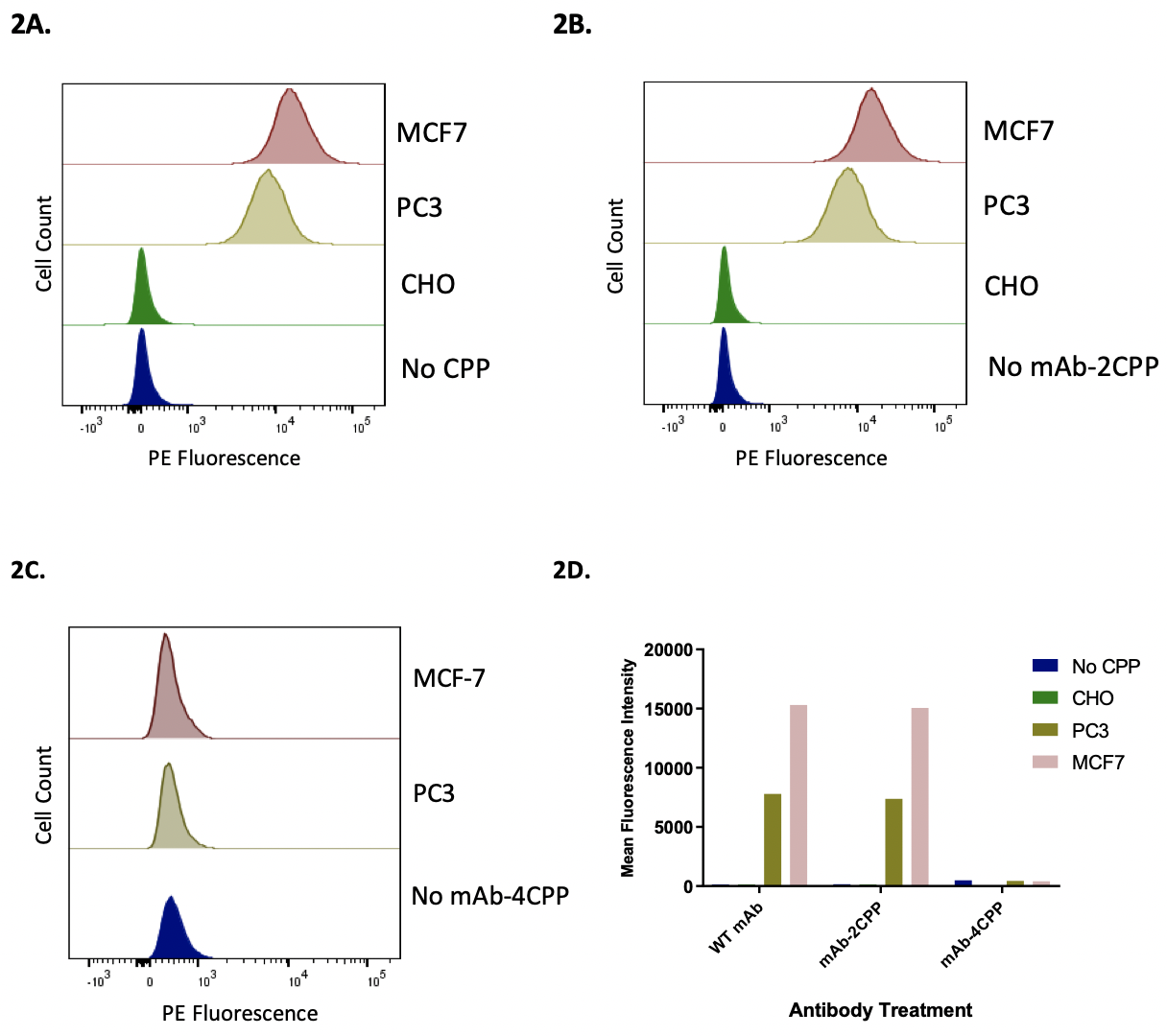
Figure 2. Flow cytometry analysis to compare the binding of wild-type and the recombinant antibodies to MCF7 and PC3 cells expressing HER2 receptor. Flow cytometry method was used to measure the binding of wild type and two recombinant antibodies, mAb-2CPP and mAb-4CPP, to MCF7, PC3, and CHO cells is measured below. Figure 2A. MCF-7, PC3, and CHO cells were stained by wild-type antibody. The wild-type antibody bound to MCF7 and PC3 cells, with the binding to PC3 cells was lower than that to MCF-7 cells. However, the wild-type antibody did not bind to CHO cells. Figure 2B. MCF-7, PC3, and CHO cells were stained by mAb-2CPP. The mAb-2CPP bound MCF7 and PC3 cells, with the binding to PC3 cells lower than that to MCF-7 cells. However, mAb-2CPP did not bind to CHO cells. Figure 2C. MCF-7 and PC3 cells were stained by mAb-4CPP. The binding of mAB-4CPP to HER2 receptor on both MCF-7 or PC3 cells was significantly impaired. Figure 2D. The binding activity in flow cytometry analysis was represented by a median assay signal.
Flow Cytometry to Characterize the Specific Binding of Wild-type Antibody and mAb-CPP Fusion Proteins to HER2 Receptor on MCF-7 and PC3 Cells
The ability of wild-type and recombinant antibodies to bind to the cell surface HER2 receptor was characterized by flow cytometry analysis. Binding is indicated by comparing the peak shift to the background control group where there was no addition of the antibody under investigation (Fig. 2). The mAb-2CPP completely preserved the HER2-binding activity. The recombinant antibody bound to MCF-7 and PC3 cells that express different levels of HER2 receptor on the surface, generating median assay signal comparable to the wild-type antibody on each tested cell line (Fig. 2A, Fig. 2B, Fig. 2D). The median binding signal is approximately two-fold higher on MCF-7 breast cancer cells than that on PC3 prostate cancer cells, consistent with a higher expression level of HER2 receptor on MCF-7. As the negative controls, both the wild-type antibody and mAb-2CPP did not bind to CHO cells that do not express HER2 receptor on the cell membrane. This further confirms that binding of the wild-type and mAb-2CPP antibodies to MCF-7 and PC3 cells are specific to the HER2 receptor. On the contrary, HER2-binding activity was significantly impaired for mAb-4CPP when an additional two copies of penetratin were fused to the N-terminus of antibody heavy chain (Fig. 2C, Fig. 2D). No binding was detected on either MCF-7 or PC3 cells. The complementarity-determining regions (CDRs) located close to the N-terminus of the antibody is responsible for antigen recognition. Incorporation of penetratin at the N-terminus of anti-HER2 mAb may have blocked its HER2 binding ability by generating steric hindrance or directly interfering with the folding of CDR structure (Chodorge et al., 2018). As a result, mAb-2CPP was used in the subsequent studies to evaluate the impact of penetratin on the internalization of recombinant anti-HER2 antibody into the HERE2 expressing cells.
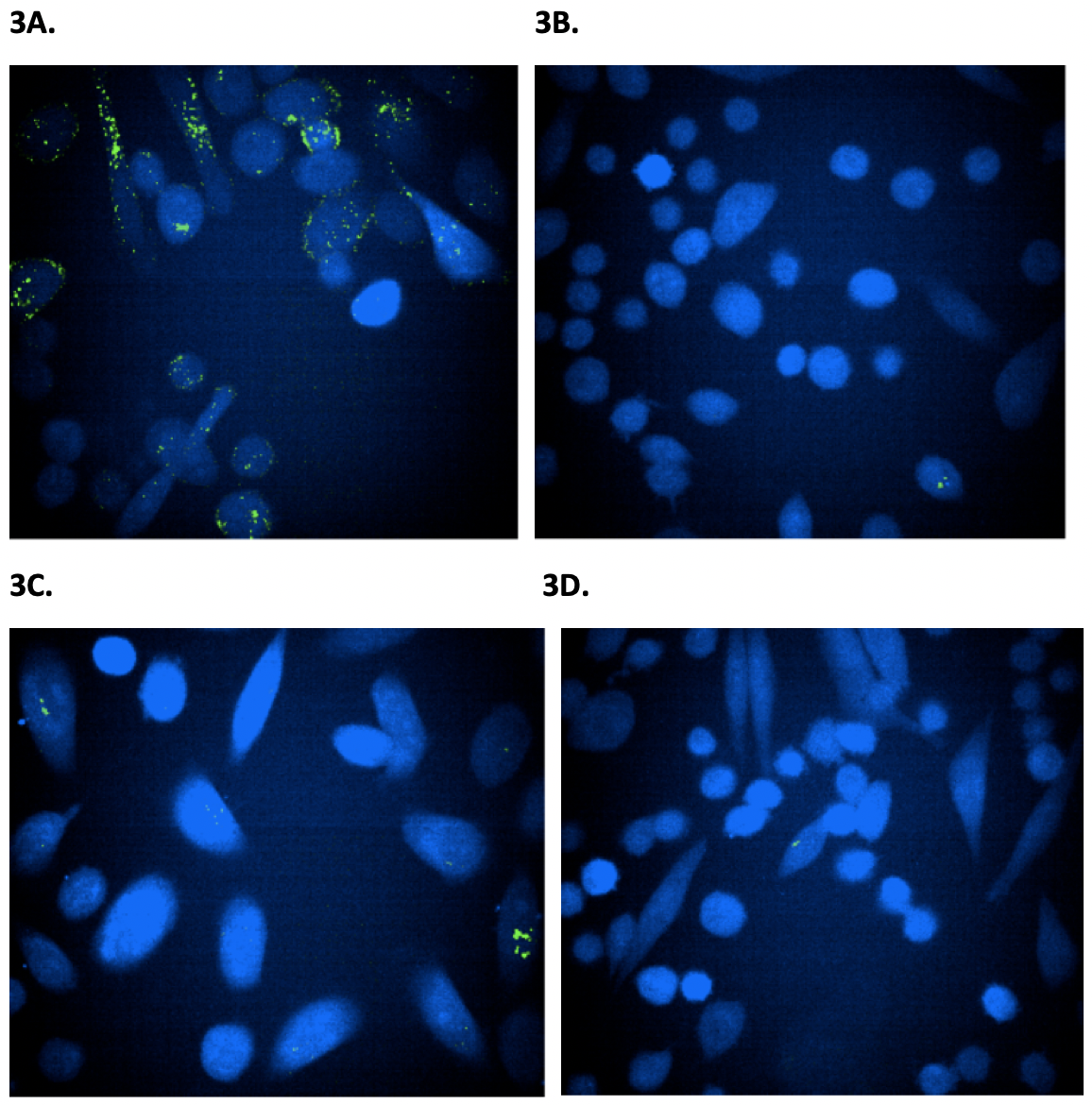
Figure 3. Confocal microscope imaging to monitor the internalization of wild-type and mAb-2CPP antibodies into PC3 and CHO cells. Confocal microscope was used to visualize the internalization of antibodies into PC3 expressing HER2 receptor and CHO cells that do not express HER2 receptor and serve as a negative control. The images were taken three hours after treating cells with antibodies. The green dots indicate internalized antibody inside cells. Figure 3A. The PC3 cells were treated with mAb-2CPP. Based on imaging, mAb-2CPP were internalized at a high level three hours after treatment. Figure 3B. The CHO cells treated with mAb-2CPP antibody showed a clean background since they do not express HER2 receptor. Figure 3C. The PC3 cells treated with wild-type antibody demonstrated a low level of internalization. Figure 3D. The CHO cells treated with wild-type antibody showed a clean background since they do not express HER2 receptor. then removed and replaced by fresh medium. Cells were then imaged under a fluorescent microscope to monitor the internalization of Alexa fluor 488 labeled antibodies at different time points (Fig. 3). MAb-2CPP was internalized as early as 3 hours after treatment, a much higher level than the wild-type antibody. (Fig. 3A, Fig. 3C). However, CHO cells showed a clean background since they do not express HER2 receptor (Fig. 3B, Fig. 3D).
mAb-2CPP Crossed the Cellular Membrane and Internalized More Efficiently Than WT mAb
The PC3 cells express a lower level of HER2 on the cell surface when compared to MCF-7 cells and were used in the immunofluorescence confocal laser scanning to image the membrane translocation activity of the wild-type and mAb-2CPP antibodies. As CHO cells do not typically express HER2 on the cell membrane, they were used in the assay as the negative control. Both PC3 and CHO cells were seeded in a 96-well plate. After overnight culture, the cells were treated by fluorescent dye, Alexa fluor 488, labeled wild-type antibody or mAb-2CPP. After removal of the media, the cells were washed once with warm DMEM media before they were stained with cell tracker to provide contrast for fluorescent antibody staining. The medium was then removed and replaced by fresh medium. Cells were then imaged under a fluorescent microscope to monitor the internalization of Alexa fluor 488 labeled antibodies at different time points (Fig. 3). MAb-2CPP was internalized as early as 3 hours after treatment, a much higher level than the wild-type antibody. (Fig. 3A, Fig. 3C). However, CHO cells showed a clean background since they do not express HER2 receptor (Fig. 3B, Fig. 3D).
The results indicate that mAb-2CPP was internalized more efficiently than the wild-type antibody. The incorpora tion of penetratin into the wild-type anti-HER antibody promotes antibody translocation to the cell membrane after it binds to the HER2 receptor on PC3 cells.
Optimize the MCF-7 and PC3 Cell Number in the 96-well Assay Plate for Sensitive CellTiter-Glo® Viability Assay after X-ray Radiation Treatment
For sensitive detection of X-ray induced cell killing, the number of MCF-7 and PC3 cells in culture needs to be op timized so that the assay signal generated in the CellTiter-Glo® viability assay will fall in the linear detection range where there is a linear relationship between the intensity of assay signal and the amount of ATP or viable cells in the culture. Furthermore, X-ray radiation would damage DNA inside living cells, which may go through a repair mechanism for recovery, generating a delay in cell death. To accurately assess the damage after X-ray treatment, we monitored the cell viability one day and three days after exposure. The optimal number of cells seeded in culture plate would ensure the assay signal measured at these time points would fall within the linear detection range.
To determine the linear detection range for both cell lines, an increasing amount of MCF-7 or PC3 cells, ranging from 97 to 25,000 cells per well, were seeded in a 96-well cell culture plate. After Day 0 (24 hours after cell seeding when the cells will be exposed to X-ray radiation), the number of viable cells in each well was assessed by CellTiter-Glo® viability assay on Day 1 (48 hours after cell seeding and 24 hours after the planned X-ray treatment) and Day 3 (72 hours after cell seeding and 48 hours after the planned X-ray treatment).

Figure 4. The growth kinetic curves for MCF-7 and PC3 cells. The CellTiter-Glo® viability assay was performed on Day 1 (48 hours after cell seeding) and Day 3 (72 hours after cell seeding) for both MCF-7 and PC3 cells. The chemiluminescent assay signal at each time point (Day 1 or Day 3) was plotted against the number of cells initially seeded to generate the cell growth kinetic curves. Figure 4A. The MCF-7 cells were seeded at 2000 cells/well so that the assay signal would fall in the mid-point of the linear range of detection in both Day 1 and Day 3. Figure 4B. The PC3 cells would be seeded at 3000 cells/well so that the assay signal would fall in mid-point of the linear range of detection. The luminescent signal generated during the assay was measured by the luminometer and recorded in relative light units (RLU).
The kinetics of cell growth were assessed by plotting the assay signal at each time point against the cell number initially seeded in assay wells. The results show that the assay signal on Day 3 was higher than that from Day 1. This was due to cell proliferation and the increased cell number as culture time elongated in the 37°C CO2 incubator. According to the kinetic curves (Fig. 4), MCF-7 cells would be seeded at 2000 cells/well (Fig. 4A) and PC3 cells at 3000 cells/well (Fig. 4B) to generate assay signals in the mid-point of linear detection range in both Day 1 and Day 3. This would allow sensitive viable cell quantification after X-ray radiation treatment.
Optimization of the Dose of X-Ray Radiation for Cell Treatment
The dose of X-ray radiation treatment should be optimized to allow the detection of difference in cell viability when the amount of GNPs in the cells varied. MCF-7 and PC3 cells were seeded in 96-well cell culture plates at 2000 cells/well and 3000 cells/well respectively. After overnight culture (Day 0), the cells were exposed to increasing doses of X-ray radiation (0 Gray to 1250 Gray). The cell viability under different doses of radiation was measured on Day 1 and Day 3. The doses that generated approximately 20% (EC20) and 50% (EC50) of the maximal cell mortality were used for radiation treatment of MCF-7 and PC3 cells. The radiation dose at EC50 was implemented to generate adequate cell eradication, while the relatively lower radiation dose at EC20 was used to investigate whether the efficacy of X-ray radiation in cell elimination would increase even at a low dose after the cancer cells uptake GNPs. The formula used for computing percent mortality (% Mortality) is defined by the following equation:

The dose-response curves were generated by plotting the % Mortality against the corresponding dose of X-ray radiation (Fig. 5). The dose-response curves were similar for MCF-7 and PC3 cells in both Day 1 (Fig. 5A) and Day 3 (Fig. 5B). The results indicate that 10 Gray and 250 Gray generated approximately 20% and 50% cell mortality respectively and would be the selected doses for X-ray radiation treatment. The use of 10 Gray would help to determine whether the presence of GNPs in the cells would generate cell death at a lower dose of X-ray radiation and thereby augment the efficacy of the cancer radiotherapy.

Figure 5. The dose-response curves of X-ray radiation. The dose-response curves were generated by plotting %Mortality against the corresponding dose of X-ray radiation in Day 1 or Day 3. The results showed that 10 Gray and 250 Gray generated approximately 20% and 50% cell mortality respectively on Day 1 and Day 3 and would be the selected doses for X-ray radiation treatment. Figure 5A. Day 1 dose-response of MCF7 and PC3 cell viability to X-ray radiation. Figure 5B. Day 3 dose-response of MCF7 and PC3 cell viability to X-ray radiation.
Reduced Cell Viability in MCF-7 and PC3 Cells After Treatment with mAb-2CPP-GNPs and Exposure to X-Ray Radiation
The wild-type mAb and mAb-2CPP were linked to GNPs (mAb-GNP and mAb-2CPP-GNP respectively) through metal-chelation at the histidine-rich metal-binding site in the antibody Fc region. This oriented conjugation method ensures that the antibody would maintain the appropriate orientation and biological activity to bind its target with high efficiency. The conjugation procedure allows a high-efficiency immobilization of antibodies on chelate-modified GNPs, achieving a protein monolayer with approximately 20 molecules per nanoparticle.

Both MCF-7 and PC3 cells were seeded in six replicate plates and treated in Day 0 for six hours with 0.3 µg/mL or 1 µg/mL of mAb-GNPs, mAb-2CPP-GNPs, and GNPs (equivalent amount), as well as wild-type mAb or mAb-2CPP controls. The two plates were then exposed to 0 Gray, 10 Gray or 250 Gray of X-ray radiation. CellTiter-Glo® viability assay was performed for one set of plates exposed to 0 Gray, 10 Gray or 250 Gray of X-ray radiation on Day 1. Subsequently, the same viability assay was performed for the remaining plates at 0 Gray, 10 Gray and 250 Gray on Day 3. Two trials were conducted with a consistent trend. Since there was a subtle difference in the concentration of mAb-GNP and mAb-2CPP-GNP, the data from one trial was reported for discussion. The results show that mAb-2CPP was not especially toxic to the cells when compared to the “Media Only” and “Media + WT mAb” control groups (Table 1). However, as reported previously in other studies (Gamberi, 2013), GNPs were cytotoxic even in the absence of X-ray radiation treatment and caused cell death in a dose-dependent manner. Therefore, it is important to target GNPs to cancer cells and minimize its cytotoxicity to healthy tissues.

GNP uptake was assessed in cells that were not exposed to X-ray radiation based on its inherent cytotoxicity (Table 2, Fig. 6). The data from a lower dose of antibody-GNP complexes at 0.3µg/mL was used for comparison among the control and variable groups. The MAb-2CPP-GNP complexes resulted in up to 50% higher cell mortality rate likely due to the delivery of more GNPs into MCF-7 or PC3 cells by mAb-2CPP. This was more prominent 24 hours (Day 1) after treatment.

Figure 6. Schematic diagram illustrating increased cell mortality rate after treatment by mAb-2CPP-GNP complexes. The mortality rate was compared between cells treated by the WT-mAb-GNPs or mAb-2CPP-GNPs. The % Increase was computed using the formula: %Increase = [(Mortality Rate_mAb-2CPP-GNP/Mortality Rate_mAb-GNP)-1] x 100%.
The results indicate that both mAb-GNP and mAb-2CPP-GNP conjugates caused mortality in MCF-7 and PC3 cells in a dose-dependent manner after X-ray radiation treatment (Table 3, Fig. 7). In other words, the mortality rate increased as a higher concentration of mAb-GNP or mAb-2CPP-GNP was used to treat MCF-7 or PC3 cells (1 µg/mL versus 0.3 µg/mL). This strongly indicates that it was the presence of GNPs that significantly enhanced cell mortality rate and the efficacy of radiation treatment. It is worth mentioning that the mortality was also observed after mAb-GNP treatment. Previous work indicates that anti-HER2 antibody can bind to HER2 receptor and internalize into cells (Rudnick et al., 2011). This would generate the background for the experiment and make the impact of penetratin on antibody inter nalization less prominent.

For both MCF-7 and PC3 cells, the cell mortality rate also increased as the dose of X-ray radiation treatment was elevated from 10 Gray to 250 Gray. The cell killing effect was observed 24 hours after treatment and was found to further potentiate up to 72 hours for each given dose of radiation. The comparison of the data also suggests that mAb-2CPP-GNP tended to generate a higher cell mortality rate than mAb-GNP, especially at 10 Gray radiation dose and 24 hours (Day 1) after X-ray exposure (Table 3, Fig. 7).
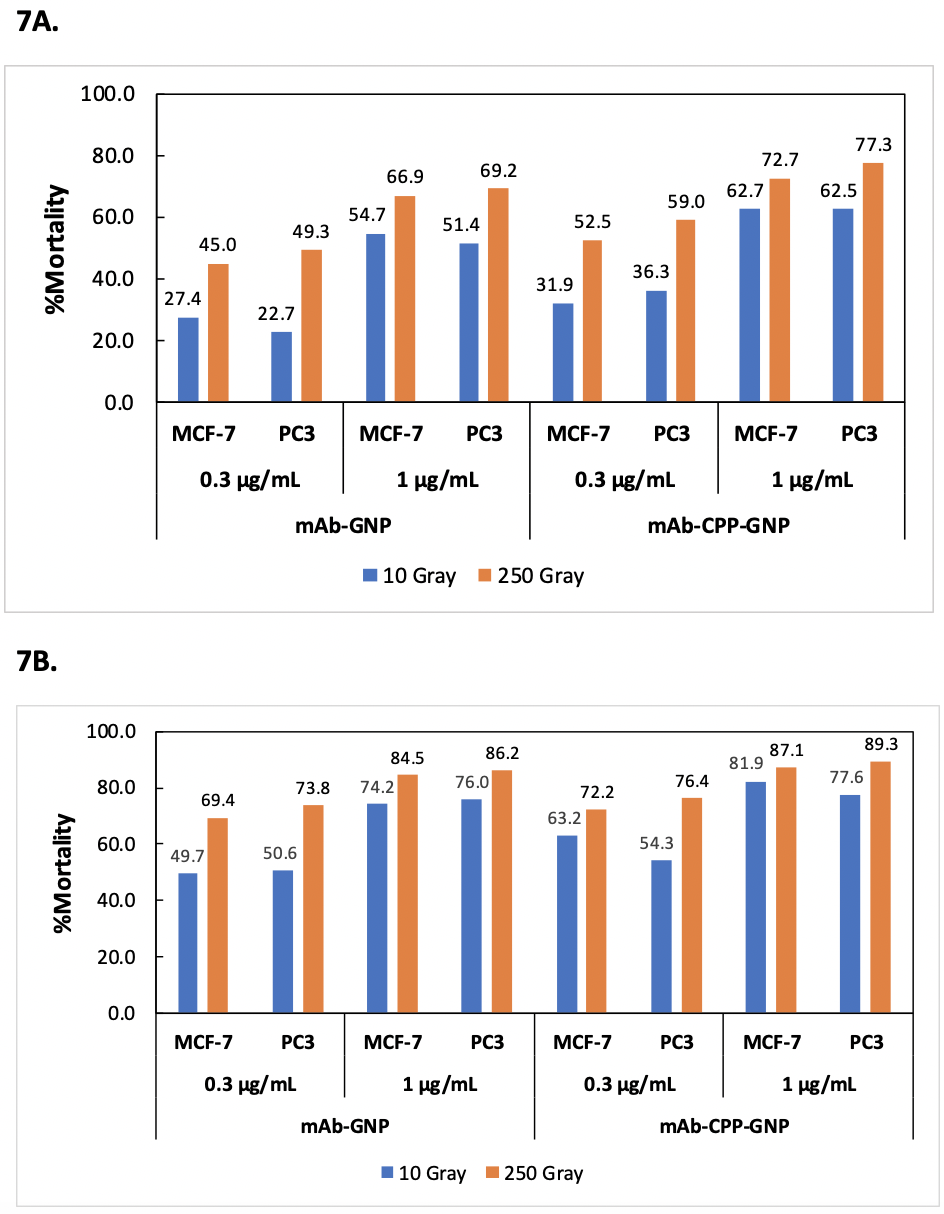
Figure 7. Schematic diagram illustrating the dose-dependent increase of cell mortality rate in response to antibody-GNP treatment and X-ray radiation. Figure 7A. Dose-dependent increase of cell mortality rate 24 hours (Day 1) after X-ray radiation exposure. Figure 7B. Dose-dependent increase of cell mortality rate 72 hours (Day 3) after X-ray radiation exposure.
Furthermore, in the presence of GNPs, a much lower radiation dose could be used to achieve a similar cancer cell mortality rate (Table 3). For example, when 1 µg/mL of mAb-2CPP-GNP and 10 Gray of radiation were used to treat MCF-7 cells, an 81.9% mortality rate was generated 72 hours after exposure to radiation. This is comparable to the 87.1% mortality rate generated by 250 Gray X-ray radiation. The result indicates that GNPs could potentially augment the efficacy of X-ray radiation and enable the use of a lower dose radiation that would generate a similar efficacy to eradicate cancer cells.
MAb-2CPP-GNPs Consistently Generated a Higher Cell Mortality Rate than Wild-type MAb-GNPs in a Statistically Significant Manner
The statistical analysis used a generalized mixed effect model specifying four main effects (test sample, sample concentration, time and radiation dose) and all possible two-way interactions of the four factors. The model also includes a random effect that accounts for the between-cell variability. The between-treatment comparisons were made in terms of the odds ratios as defined by the formula:

The two-sided statistical tests conclude that mAb-2CPP-GNPs consistently generated a higher cell mortality rate under all testing conditions than wild-type mAb-GNPs (p value < 0.05) (Fig. 8). The difference was most prominent when 1 µg/mL of mAb-2CPP was used to treat cells followed by subsequence exposure to 10 Gray of X-ray radiation.
DISCUSSION
A protein engineering method was implemented to fuse 2 copies of penetratin (CPP) to the C-terminus of the anti-HER2 antibody heavy chain. The CPP-mAb fusion protein completely preserved therapeutic antibody binding activity. However, the fusion of CPP to both the N-terminus and C-terminus of heavy chain abolished the antibody binding activity. These results could be used to guide the creation of other recombinant cell penetrating mAbs. MAb-2CPP carrying two copies of CPP was able to promote the internalization of anti-HER2 antibody into MCF-7 and PC3 cells expressing HER2 receptor. The ability to engineer a cell penetrating antibody would allow the antibody to target intracellular pathogenesis-related protein and expand its potential for therapeutic applications.
X-ray radiation is commonly used in the medical industry and known for high penetration depth and high energy in individual photons (Gianfaldoni et al., 2017). Its interaction with GNPs was investigated in this study, indicating that X-ray could serve as the source of high energy photon to power GNP. The results demonstrate that GNPs generated synergistic effects with X-ray radiation to significantly enhance its cell killing effect. Cells treated by GNPs or antibody-GNP complexes effectively responded to X-ray radiation in a dose-dependent manner. The cell mortality rate increased with a higher concentration of mAb-GNP or mAb-2CPP-GNP used for cell treatment or an elevated level of X-ray radiation. The efficacy of radiation was observed 24 hours after treatment and further potentiated up to 72 hours after radiation treatment. In addition, GNPs augmented the effective dose of X-ray radiation; a lower radiation dose could be used to achieve a similar cancer cell mortality rate, thus reducing the side effects and decreasing the risks to patients.
Based on the results from fluorescence imaging and the cell uptake assay, mAb-2CPP-GNP was able to shuttle more GNPs into MCF-7 and PC3 cells, consistently leading to a higher cell mortality rate than mAb-GNPs in a statistically significant manner. This effect would have been more prominent if an antibody with no cell internalization capacity could be evaluated in the study.
In addition to penetratin, which was investigated in the current study, more CPPs could be screened and evaluated for their capacity to promote the internalization of antibodies that otherwise do not cross the cell membrane effectively. When available, different sizes of GNPs could also be compared for their cell uptake and cytotoxicity after exposure to radiation. An optimized antibody-GNP complex could be tested in melanoma or other cancer types in an animal model to further assess the feasibility of GNPs to augment the efficacy of X-ray radiation mediated cancer heat therapy (Letfullin et al., 2016; Paro et al., 2017).
ACKNOWLEDGMENTS
I would like to express my sincere appreciation to Rebecca Hawkins for her valuable and constructive suggestions during the planning and development of this research work. I would also like to thank Dr. Qianqiu Li for her help with the statistical analysis.
REFERENCES
Ali, S. A., Teow, S., Omar, T. C., Khoo, A. S., Choon, T. S. and Yusoff, N. M. (2016.) A cell internalizing antibody targeting capsid protein (p24) inhibits the replication of HIV-1 in T Cells lines and PBMCs: A proof of concept study. PLoS ONE, 11(1): e0145986.
Chodorge, M., Celeste, A. J., Grimsby, J., Konkar, A., Davidsson, P., Fair-man, D., Jenkinson, L., Naylor, J., White, N., Seaman, J. C., Dick-son, K., Kemp, B., Spooner, J., Rossy, E., Hornigold, D. C., Trevaskis, J. L., Bond, N. J., London, T. B., Buchanan, A., Vaughan, T., Rondinone, C. M., Osbourn, J. K. (2018) Engineering of a GLP-1 analogue peptide/anti-PCSK9 antibody fusion for type 2 diabetes treatment. Nature, 8:17545.
Dinca, A., Chien, W-M., Chin, L. and Chin, M. (2016). Intracellular delivery of proteins with cell-penetrating peptides for therapeutic uses in hu-man disease. International Journal of Molecular Sciences, 17, 263-276.
Dupont, E., Prochiantz, A. and Joliot, A. (2015). Penetratin story: an over-view. Cell-penetrating peptides: Methods and Protocols, Methods in Molecular Biology, 683, doi:10.1007/978-1-60761-919-2_2, 21-29.
Fay, F. and Scott, C. J. (2011). Antibody-targeted nanoparticles for cancer therapy. Immunotherapy, 3(3), 381-394.
Gamberi, T., Magherini, F., Bini, L., Messori, L., Gabbiani, C., Pietrovito, L., Modesti, P. A. and Modesti, A. (2013). New insights into the molecular mechanisms of selected anticancer metal compounds through bioinformatic analysis of proteomic data. Journal of Proteomics and Bioinformatics, S6, 2.
Gianfaldoni, S., Gianfaldoni, R., Wollina, U., Lotti, J., Tchernev, G. and Lot-ti, T. (2017). An Overview on Radiotherapy: From Its History to Its Current Applications in Dermatology. Journal of Medical Sciences, 5(4), 521–525.
Hu, C., Niestroj, M., Yuan, D., Chang, S. and Chen, J. (2015). Treating cancer stem cells and cancer metastasis using glucose-coated gold nanoparticles. International Journal of Nanomedicine, 10, 2065–2077.
Jain, S., Hirst, D. G. and O’sullivan, J. M. (2012). Gold nanoparticles as novel agents for cancer therapy. The British Journal of Radiology, 85(1010), 101-113.
Kaur, P., Aliru, M. L., Chadha, A. S., Asea, A. and Krishnan, S. (2016). Hyperthermia Using Nanoparticles – Promises and Pitfalls. International Journal of Hyperthermia, 32(1), 76–88.
Lim, K. J., Sung, B. H., Shin, J. R., Lee, Y. W., Kim, D. J., Yang, K. S., and Kim, S. C. (2013) A Cancer Specific Cell-Penetrating Peptide, BR2, for the Efficient Delivery of an scFv into Cancer Cells. PLoS ONE, 8(6): e66084-e.
Letfullin, R. R, Rice, C. E. W. and George, T. F. (2016). Nanoparticle-enhanced X-ray therapy for cancer. SPIE Digital Library.
Mazzucchelli, S., Truffi, M., Fiandra, L., Sorrentino, L., and Corsi, F. (2014). Targeted approaches for HER2 breast cancer therapy: News from nanomedicine? World J Pharmacology, 3(4): 72-85.
Paro, A. D., Shanmugam, I., and van de Ven, A. L. (2017). Nanoparticle-Mediated X-Ray Radiation Enhancement for Cancer Therapy. Methods of Molecular Biology, 1530: 391–401.
Rudnick, S. I., Lou, J., Shaller, C. C., Tang, Y., Klein-Szanto, A. J. P., Weiner, L. M., Marks, J. D., and Adams, G. P. (2011). Influence of affinity and antigen internalization on the uptake and penetration of anti-HER2 antibodies in solid tumors. Cancer Research, 71(6), 2250–2259.
Saha, S., Xiong, X., Chakraborty, P. K., Shameer, K., Arvizo, R. R., Kudgus, R. A., Dwivedi, S. K. D., Hossen, N., Gillies, E. M., Robertson, J. D., Dudle, J. T., Urrutia, R. A., Postier, R. G., Bhattacharya, R., and Mukherjee, P. (2016). Gold nanoparticle reprograms pancreatic tumor microenvironment and inhibits tumor growth. ACS Nano, 10, 12, 10636-1065.
Watson, G. M., Kulkarni, K., Brandt, R., Del Borgo, M. P., Aguilar, M., and Wilce, J. A. (2017). Shortened penetratin cell-penetrating peptide is insufficient for cytosolic delivery of a Grb7 targeting peptide. ACS Omega, 2, 670−677.
Zhong, X., Ma, W., Meade, C. L., Tam, A. S., Llewellyn, E., Cornell, R., Cote, K., Scarcelli, J. J., Marshall, J. K., Tzvetkova, B., Figueroa, B., DiNino, D., Sievers, A., Lee, C., Guo, J., Mahan, E., Francis, C., Lam, K., D’Antona, A. M., Zollner, R., Zhu, H. L., Kriz, R., Somers, W., and Lin L. (2019).)Transient CHO expression platform for robust antibody production and its enhanced N-glycan sialylation on therapeutic glycoproteins. Biotechnol. Prog., 35 (1), 1-12.