
Author: Maureen A. Carey & Eric S. Ho
Institution: [1] Department of Microbiology, Immunology, and Cancer Biology, University of Virginia, Charlottesville, VA 22908, USA [2] Department of Biology with affiliation to Computer Sciences, Lafayette College, Easton, PA 18042
doi:10.22186/jyi.33.1.28-41
ABSTRACT
Lyme disease is the most common vector-borne disease in the United States and is typically caused by the bacterium Borrelia burgdorferi. Although often curable, delayed diagnosis due to nonspecific symptoms risks systemic complications, and some patients experience symptoms despite bacterial clearance from the body. We hypothesized that B. burgdorferi infection induces a self-perpetuating cascade of immunological responses such that symptoms remain after infection or causes residual damage to patients’ immune system and tissues. We present a transcriptome study of B. burgdorferi infection in murine heart and brain tissues using the Next Generation Sequencing technology and computational methods to identify differentially expressed genes, particularly for evidence of active inflammatory pathways. Our results reveal differential expression of five genes in an infected heart. These differentially expressed genes are enriched in pathways related to immune functions in heart tissue. Our study indicated that B. burgdorferi infection triggers immune response pathways similar to other pathogens, and some genes were found to be unique to infection by B. burdorferi, suggesting the potential for development of specific therapeutic targets to treat B. burgdorferi infection. In the brain, 66 genes were differentially expressed. These genes were enriched in pathways that facilitate the pathogen’s crossing of the blood-brain barrier. Although the mouse model of B. burgdorferi infection fails to recapitulate human neuroborreliosis, we observed damage to the integrity of the blood-brain barrier upon peripheral infection. This study elucidates mechanism of infection unique to Borrelia and clarifies the role of a mouse model of Lyme disease.
INTRODUCTION
Lyme disease is prevalent from southern Scandinavia to the northern Mediterranean countries and in the northeastern United States (U.S.). In the U.S., Lyme disease is the most common vector-borne disease: over 251,000 cases were reported between 2005 and 2014, with about 25,000 confirmed cases each year. Most cases occur in the northeast; however, notable expansion was observed in the Great Lakes region (CDC 2014). Lyme disease is caused by the infection of Borrelia burgdorferi sensu lato (family Spirochaetaceae), a diderm, microaerophilic spirochete bacteria (Wang et al., 1999). Within the genus Borrelia, three other species (B. afzelii, B. garinii, and possibly B. valaisiana) can cause the disease, but are more prevalent on the European continent (WHO, 2006). Other Borrelia species are carried by soft-bodied ticks and cause relapsing fevers (Garcia-Monco et al., 1997). All four pathogenic species of Borrelia are spread to humans by the bite of an infected tick. In the U.S., two blacklegged, or deer, tick species (Ixodes scapularis and Ixodes pacificus) are known to carry B. burgdorferi. The bacteria infect several mammal and bird species and are transmitted during the tick’s blood meals (Rosa et al., 2005).
Although Lyme disease is usually curable with prompt antibiotic treatment, nonspecific symptoms make early diagnosis difficult, and untreated infection can induce rheumatic, cardiac, and neurologic complications. The current screening test is still suboptimal in detecting Lyme reliably (Centers for Disease, & Prevention, 1995; Dressler et al., 1993). Lyme is often diagnosed after the emergence of the classic bulls-eye-shaped rash at the site of the tick bite, which occurs in over 70% of patients (McConville, 2014). The infection spreads throughout the body, causing general inflammation during the early dissemination stage, and years after initial infection, painful arthritis and joint swelling are observed among 60% of patients (McConville, 2014). Borrelia are transported throughout the body, and persistent infections are established in the skin, joint, heart, bladder, and, in only humans and primates, the central nervous system (Rosa et al., 2005).
Some of these tissues are particularly affected by infection-induced inflammation. Lyme carditis (inflammation of the heart tissue, interfering with its electrical activity) occurs in 4-10% of infections during the early dissemination stage. Carditis responds well to antibiotic treatment; however, because it occurs so early in the infection process and Lyme disease is difficult to diagnose, it can be fatal (McAlister et al., 1989). Additionally, 10-15% of Lyme disease cases manifest neurological conditions, such as pain caused by temporary or permanent inflammation of the nerves, meningitis, memory and anxiety problems, depression, and both cranial and peripheral neuritis (Narasimhan et al., 2003; Pachner, & Steere, 1984; Rupprecht et al., 2008).
Some patients will experience Post-Treatment Lyme Disease Syndrome (PTLDS), a chronic manifestation of Lyme disease. PTLDS is diagnosed when symptoms continue despite bacterial clearance from the body (McConville 2014). Unlike many other gram-negative bacteria, little epidemiological evidence shows antibiotic-resistant Borrelia infections to be a threat; however, the prevalence of persistent symptoms is concerning. Because antibiotic treatment does not necessarily resolve PTLDS, understanding how Borrelia affects the body, especially the heart and brain tissues, is crucial in reducing the burden of this disease. We hypothesize that Borrelia infection induces a self-perpetuating cascade of immunological responses, such that symptoms remain after infection. The B31 B. burgdorferi genome has been fully sequenced, consisting a small linear chromosome (~900kb) and 21 unique plasmids (5-56kb) (Fraser et al., 1997), but does not reveal any obvious virulent elements (Rosa et al., 2005). Thus, looking at the transcriptional activities of the infected host rather than the genome of B31 B. burgdorferi may shed light the immune response and on its pathogenesis.
A microarray study of Borrelia genes during infection of heart and CNS tissue in non-human primates revealed elevated expression of over 90 genes in bacteria in the CNS when compared to bacteria in the heart (Narasimhan et al., 2003), indicating that parasite-host responses are different in the two tissues. Infection induces a macrophage response and upregulated cytokine expression in the murine macrophage cell line (Wang et al., 2008). However, little is know about the host’s transcriptional response at tissue level upon Borrelia infection. Here, we present a transcriptome study that integrates experimental and computational methods to probe for the effect of B. burgdorferi infection on gene expression, and subsequently, biological pathways of inflammation in murine heart and brain tissues. We have designed a dual-method redundant pipeline to overcome issues arising from the lack of replicates owing to the scarcity of samples and the high cost of RNA-sequencing (RNA-seq). This method will allow us to better study and characterize acute and persistent Borrelia infection.
MATERIALS AND METHODS
Culture and Infection
B31-MI B. burgdorferi, from ATCC (Manassas, VA), was grown in BSK-H (Sigma BSK-H Complete, St. Louis, MO) at 37°C to a concentration of 7.2x107 viable spirochetes/mL at the Baumgarth lab at University of California Davis and shipped on ice for next-day infections.
Six female C3H/HeJ mice (The Jackson Laboratory, Bar Harbor, ME), aged 6-8 weeks old, were infected, and four female C3H/HeJ mice, also aged 6-8 weeks old, were used as controls. Two injections of approximately 0.5mL each were injected into each mouse subcutaneously in the mid-back with a 21-gauge needle. Control mice were injected with BSK-H media via the same protocol. C3H/HeJ mice carry a chromosomal inversion on Chromosome 6 (Chang, 2015), which yields no phenotypic change, as well as mutations in the Pde6b and Tlr4 genes. The Pde6b mutation causes retinal degeneration and eventual blindness. The Tlr4 mutation makes these mice more tolerant to endotoxin in bacterial infections. Higher than the minimum dose (3.6x105 times higher) (Barthold et al., 1993; Rego et al., 2014) of spirochetes was injected to the mice to ensure infection. Arthritic swelling was observed in all three experimental mice collected on day 14, and in one mouse collected on day 42. All mice were used in accordance with Lafayette College's Institutional Animal Care and Use Committee approved protocol that followed the guidelines for ethical conduct in care and use of animals.
RNA Extraction
Only mice infected for 14 days were selected for RNA-seq to explore the acute phase of the disease. They were sacrificed with carbon dioxide gas and then cervical dislocation. Samples of heart and brain tissue were collected at 14 and 42 days. RNA extraction using TRIzol Reagent (Ambion, Austin, TX) was conducted, following the manufacturer’s protocol, from sample tissues. A preliminary analysis of one sample from control and experiment was conducted. Both samples, brain and heart, were extracted from control and experimental animals after 14 days of infection. These samples were chosen as a condition-constant (day) control-experimental pair for their high concentrations and 260/280 ratios indicative of higher RNA purity (Table S1).
RNA-Seq
One sample was selected from control and experimental conditions from heart and brain tissues for RNA-seq. Each sample consisted of 5ug of poly(A)+ total RNA. Single-end RNA-seq was performed in Illumina HiSeq platform offsite by GENEWIZ (GENEWIZ 2013). Sequencing results were returned in FASTQ files in which short read was about 50 bps long on average. The total number of short reads ranged from 47 million to 59 million per sample. The quality of short reads was checked by FASTQC (Andrews); average Q-score was 37 and over 94% of the short reads was above 30 (Table S2).
Differentially Expressed Gene Analysis
We built a dual, redundant pipeline to circumvent the scarcity of replicates, in which each dataset was processed twice by two principally distinct methods. The advantages of this pipeline include the elimination of method bias and the confidence of identifying truly differentially expressed genes (DEGs).
DEGs were identified largely by the Tuxedo pipeline (Trapnell et al., 2012) with some modifications (Figure 1A-B). Before making DEG calls, short reads obtained from Next Generation Sequencing (NGS) were mapped to the mouse genome (mm10 (Browser)) by Tophat (v2.0.10) (Trapnell et al., 2009) via alignment engine bowtie2 (v2.2.1.0) (Langmead, & Salzberg 2012). Following short reads mapping was the assembly of overlapping short reads into long transcripts. By counting the number of transcripts mapped to genes, gene expression levels were determined.
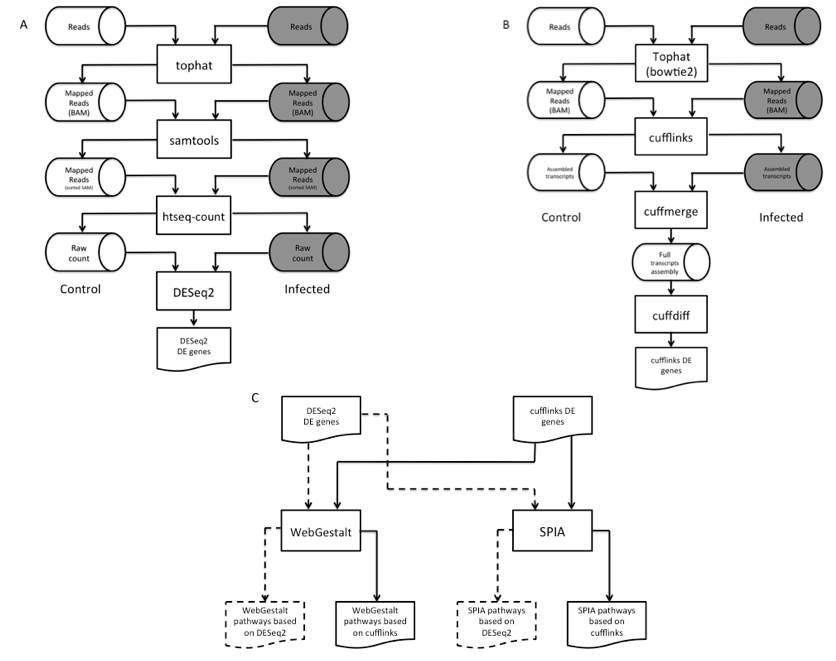
Figure 1. Workflow of the dual-method approach to differential gene expression analysis and signaling pathway identification. (A) Short reads mapping and differentially expressed gene (DEG) identification using DESeq2. (B) RNA-seq short reads mapping and DEG identification using the Tuxedo pipeline. (C) Signaling pathway analysis of DEGs using WebGestalt and SPIA.
Two DEGs callers were used in our dual-method pipeline: cufflinks (v2.1.1) (Trapnell et al., 2010) and DESeq2 (v3.2.1) (Love et al., 2014). Figure 1A illustrates the overview of the DESeq2 pipeto line. A slight alteration was done the Tuxedo pipeline in which the number of transcripts mapped to each gene (raw count) was prepared by htseq-count (v0.6.1p1) (Anders et al., 2015) per sample before running DESeq2. This pipeline identified 365 (p < 0.10) and 168 (p < .04) DEGs in heart and brain, respectively (Supplemental File 2, Tables S4 & S6). To corroborate with the DEGs found by DESeq2, we also used the standard Tuxedo cufflinks package as an alternative method to analyze the genome-wide gene expression levels between the two conditions. This package comprises of three programs, namely cufflinks, cuffmerge, and cuffdiff. The result is a list of DEGs that show statistical significance expression patterns between control and infection.
For quality assurance purpose, RNA-seq and DEG results were inspected by a visualization method CummeRbund (v2.10.0), an R package (cummeRbund). Bias in harvesting RNA samples from control and experimental conditions may cause misleading conclusion in gene differential expression analysis. Thus, we used CummeRbund to reveal genome-wide expression distribution plots under two conditions of two tissues. Figure S1 shows similar distributions of genes in control and experimental conditions, meaning the absence of sequencing bias among our samples and both control and experimental mice of each tissue type had a similar quantity of total reads on a genome scale. Thus, expression levels are comparable on locus-focused basis.
CummeRbund also generates scatterplot to show the widespread of DEGs in experiments. Scatterplots of differential gene expression (Figures S2 and S3) showed the presence of a small set of differentially expressed genes between the two conditions in the brain and heart tissues. By using cuffdiff, 136 and 100 genes in heart tissue and brain tissue were discovered to express differentially, respectively (Supplemental File 1, Tables S3 & 35).
Signaling Pathway Analysis
Similar to the discovery of DEGs, two distinct signaling pathway analysis tools were used to search for biological pathways perturbed by DEGs due to B. burgdorferi infection: WebGestalt (Zhang et al., 2005), and SPIA (v3.2.1) (Tarca, 2013). Both methods sourced biological pathway information from KEGG pathway database (Kanehisa, & Goto, 2000; Kanehisa et al., 2014). WebGestalt detects enriched pathways by identifying over-represented Gene Ontology (GO) (Ashburner et al., 2000) terms associated with DEGs. The underlying statistical test used to substantiate over-representation of GO terms is the hypergeometric test. Thus, it assumes DEGs are independent of each other. Such a condition may not hold, as DEGs belonging to the same pathway inherently interact directly or indirectly with each other.
We used SPIA to cross-examine results obtained from WebGestalt. SPIA harnesses genes’ topological relationship in assessing the degree of perturbation exerted on the network by DEGs. More DEGs in a pathway indicates greater significance that the experimental condition induced a perturbation in that pathway. The location of the gene in the pathway is also taken into consideration by SPIA. For example, insulin receptor anchored at the cell surface functions as an on/off switch in the insulin signaling pathway. Thus, its differential expression induces a larger ripple effect to the downstream cellular processes than genes situated at the end of the cascades.
In our dual-method approach, each pathway analysis tool received two lists of DEGs, one from each aforementioned DEG calling methods, and produced four lists of predicted pathways for each tissue (Figure 1C). We then overlapped these lists to reliably identify pathways perturbed by B. burgdorferi infection. Pathways found in at least three out of four lists were selected for analysis (Table 1).

Table 1. Altered heart pathways associated with differentially expressed genes (DEGs). Top ten pathways ordered by the number of DEGs are listed in below. Only nine pathways in brain tissue were identified by WebGestalt and DESeq2. pPERT is the p-value for a pathway to be perturbed by DEGs. Pathways shared by at least three datasets are in bold.

Table 2. Altered brain pathways associated with differentially expressed genes (DEGs). Top ten pathways ordered by the number of DEGs are listed in below. Only nine pathways in brain tissue were identified by WebGestalt and DESeq2. pPERT is the p-value for a pathway to be perturbed by DEGs. Pathways shared by at least three datasets are in bold.
RESULTS
Differentially Expressed Genes
Cufflinks and DESeq2 identified 136, and 365 DEGs in heart tissue, respectively (Tables S3 and S4). Surprisingly, these two sets of DEGs overlapped meagerly. We then checked whether the two sets of DEGs still pertained coherent biological functions. We grouped DEGs by molecular functions using WebGestalt’s GO Slim Classification function. This function virtually determines molecular function GO terms enrichment among genes. As seen in Figure 2A, the two sets of DEGs exhibited highly similar GO molecular function profile; 15 out of 17 molecular functions were shared between the two sets. Moreover, the order of five functions was preserved between the two profiles. These five functions are protein binding, ion binding, molecular transducer activity, transporter activity, and chromatic binding. Furthermore, we repeated the same analysis using DAVID (Huang da et al., 2009a; Huang da et al., 2009b), a similar method but independently developed by a different research group. The two sets shared the top two biological process GO terms: immune response, and defense response (Tables S7 and S8).
In brain tissue, cufflinks and DESeq2 identified 100 and 168 DEGs, respectively (Tables S5 and S6). The two sets of brain DEGs shared 67 genes or 67% of cufflinks’s predictions. Out of the 14 molecular functions of DEGs, 12 functions were common between the two groups (Figure 2B). Additionally, the two methods found the same top six functions in the two datasets but in a slightly different order. Similarly, the two sets of brain DEGs were analyzed using DAVID. Biological processes behavior and locomotive behavior were shared between the two sets (Tables S9 and S10).

Figure 2. Comparison of Gene Ontology (GO) molecular function terms. (A ) Molecular function GO terms of heart DEGs generated by DESeq2 (left) and cufflinks (right). In both panels, the numbers below the captions of the bars represent the order sorted by the number of GO terms in each group. Underlined numbers on the right panel signify the conservation of their order on both panels. (B) Molecular function GO terms of brain DEGs generated by DESeq2 (left) and cufflinks (right).
Immune Response to B. burgdorferi Infection in Heart Tissue
As genes do not function alone, the infected host is expected to launch concerted biological processes to battle against B. burgdorferi. Thus, we examined whether or not the list of DEGs originated from common pathways in response to B. burgdorferi infection. We used a dual-method approach to overcome the issue of replicate-depletion in biological pathway analysis in which four sets of predicted pathways were generated for each tissue (Figure 1C). The top ten pathways, by the highest number of DEGs with applicable p-values, were selected for analysis in this study (Table 1). Ten genes belonging to the chemokine signaling pathway were up-regulated in B. burgdorferi infection (Figure 3A). Upregulation of β-arrestin 1 (Arrb1) was detected and is upstream of a myriad of downstream factors in response to infection. This result suggests that B. burgdorferi infection activated Arrb1, which stimulated a broad inflammatory response.

Figure 3A. Biological pathways altered by B. burgdorferi infection in heart tissue. Pathway diagrams were generated by Pathview (Luo, & Brouwer 2013). (A) Chemokine signaling pathway (mmu04062).
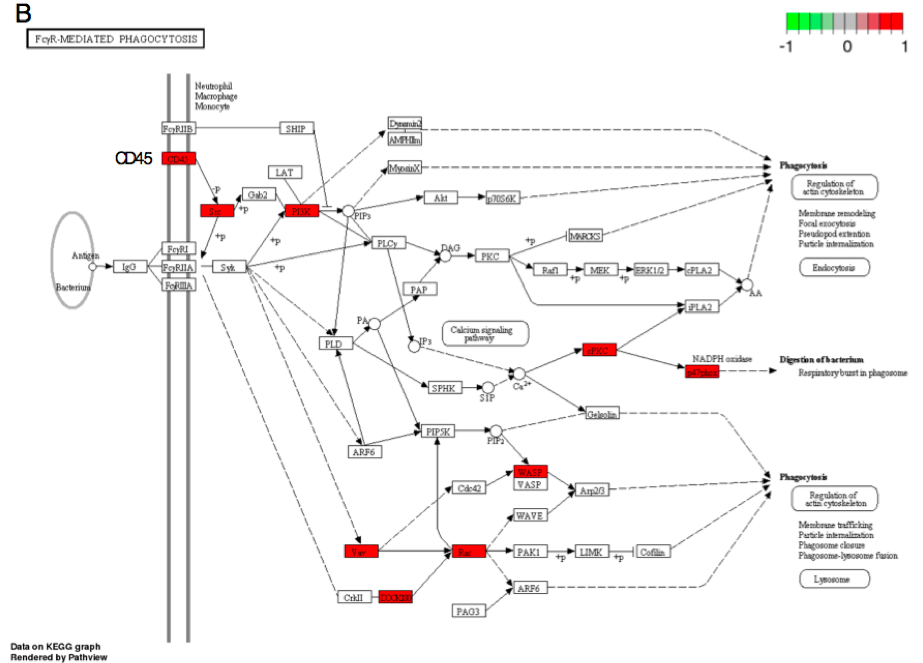
Figure 3B. FcγR-mediated phagocytosis (mmu04666).
Our enrichment results indicated that Borrelia infection in heart also perturbs the same set of genes as Staphylococcus aureus (Figure 3C) and Leishmaniasis (Figure 3E). Unlike Borrelia, Staphylococcus aureus is a gram-positive bacterium and Leishmaniasis is caused by parasites (genus Leishmania). We have found that similar to S. aureus infection (as defined by a gene cluster in DAVID), the complement genes C3 and C1Q were up-regulated in B. burgdorferi infection (Figure 3C). Similarly, three genes from the complement system (C3b, C3bi, CR3) were up-regulated by B. burgdorferi infection and Leishmaniasis (Figure 3E). The complement system supplements the activities of antibodies and aforementioned phagocytosis to remove pathogenic particles and cells.

Figure 3C. Response to S. aureus infection (mmu05150).

Figure 3D. Osteoclast differentiation (mmu04380).

Figure 3. Response to Leishmanias infection (mmu05140).
Calcium Signaling in Response to B. burgdorferi Infection in Brain Tissue
When compared with heart tissue, only three pathways in brain tissue were identified by SPIA and WebGestalt: calcium signaling, genes involved in gap junction, and melanogenesis (Table 1). Two G-Protein Coupled Receptors (GPCRs) implicated in calcium signaling, glutamate metabotropic receptor 5 (Grm5) and adenosine A2a receptor (Adora2a) showed differential expression (Figure 4A). Upregulation of Grm5 triggered a higher expression of a downstream factor PLCβ (phospholipase C beta) in the pathway. In contrast, Adora2a was down-regulated compared to heart tissue.
The second neurological pathway perturbed by B. burgdorferi infection was the gap junction (Figure 4B). The gap junction serves intercellular exchange of ions or small molecules between neighboring cells’ cytosolic compartments. Inflammatory response attributed to infection is often associated with the loss of such an exchange channel (Eugenin et al., 2012). Five genes in this pathway were perturbed, with subpathways both up- and down-regulated (Figure 4B). To our knowledge, no literature suggests an association between melanogenesis and bacterial infection. Two genes from this pathway, adenylate cyclase 4 (Adcy4) and phospholipase C beta 1 (Plcb1; Figures 4A-C), also participate in calcium signaling and gap junction pathways discussed above.

Figure 4. Biological pathways altered by B. burgdorferi infection in brain tissue. (A) Calcium signaling pathway (mmu04020).

Figure 4. Gap junction (mmu04540). Circled are Adcy4 and Plcb1 genes, discussed in text.

Figure 4C. Melanogenesis (mmu04916). Circled are Adcy4 and Plcb1 genes, discussed in text.
DISCUSSION
Differentially Expressed Genes Identification
We built a dual, redundant pipeline to identify DEGs associated with B. burgdorferi infection in heart and brain tissue, in which each dataset analyzed by two principally distinct methods (cufflinks and DESeq2) and interpreted with multiple enrichment approaches. We found hundreds of genes to be differentially expressed via the two methods in each tissue.
The set of DEGs identified in heart tissue by our two methods (Cufflinks and DESeq2) overlapped meagerly. This could be partly attributed to the difference of bowtie2 and htseq-count in mapping of non-unique short reads to genes/genome. Bowtie2 considers short reads to be mappable if the number of mismatches against genome falls below a specified threshold. This permits the possibility of double-counting non-unique reads in multiple genomic locations. Besides mismatch factor, htseq-count only counts short reads that can be mapped to a single location in the genome. Thus, fewer reads are mapped by htseq-count than bowtie2, and this difference in mapping strategy likely contributed to the difference in DEGs being identified. Intriguingly, regardless of which differentially expressed gene calling methods we used, results from WebGestalt and DAVID coherently point to the activation of immune system despite the meagerly overlapping of the two DEG lists.
Immune Response to Borrelia Infection in Heart Tissue
In heart tissue, chemokine signaling, Fc gamma R-mediated phagocytosis, osteoclast differentiation, and both S. aureus and Leishmania infection-response are associated with B. burgdorferi infection. Chemokine signaling and phagocytosis events are to be expected, as B. burgdorferi actively infects heart tissue during early stages of the disease (Armstrong et al., 1992). Our results indicated that B. burgdorferi infection activated Arrb1, which is known to stimulate a broad innate and adaptive inflammatory responses (Jiang et al., 2013). Little is known about the difference in pathogenicity between S. aureus, Leishmania, and Borrelia; however, it is not surprising to see similar genes and biological pathways are mobilized to defend the host against pathogens as many innate (early) immune responses are non-specific. Osteoclast differentiation was likely perturbed because S. aureus infects osteoblasts (osteomyelitis) (Rasigade et al., 2013; Webb et al., 2007) and associates with osteoblast differentiation pathways (Figure 3D). Although Borrelia does not infect osteoclasts, the similarities in infection response may produce this observation. The shared pathways predicted by the two distinct pathway methods unequivocally indicate upregulation of phagocytosis and pathways associated with response to infection in heart tissue. The altered gene expression indicates activation of white blood cells, induction of the complement system, and cellular targeting for immune destruction of tissue cells through alterations in receptor proteins. We observed these immunological reactions in heart only, suggesting tissue-specific targeting of B. burgdorferi primarily in the heart. However, the number of DEGs (>100) may be too extensive to draw any actionable therapeutic conclusions at this stage. Further investigation is needed to elucidate essential symptomatic genes.
Blood-Brain Barrier Disruption in Response to Borrelia Infection in Brain Tissue
Only three consensus pathways were perturbed in the brain tissue by our dual-method pipeline. Moreover, these pathways had fewer differentially expressed genes compared to the heart tissue results (Table 1). This is expected: because B. burgdorferi does not actively infect murine brain tissue (Radolf et al., 2012), a less cohesive response occurs upon host infection as a variety of cell types are responding to inflammation, not generating an inflammatory response. We observed perturbations in calcium signaling, gap junction, and melanogenesis. Calcium signaling has been shown to influence bacterial infection (Soderblom et al., 2005; TranVan Nhieu et al., 2004). We propose that this phenomenon is exploited by B. burgdorferi to cross the blood-brain barrier (Coureuil et al., 2013; Grab et al. 2005; Halperin, 2015), even if these bacteria fail to establish infection (Radolf et al., 2012) once across the barrier. This perturbation of the blood-brain barrier could be used to study human neuroborreliosis. Previous studies indicate that neurological symptoms exhibited by Borrelia infection in humans may be attributed to the success of Borrelia in crossing the blood-brain barrier and attacking the CNS (Grab et al., 2009). Our results are consistent with these findings, suggesting that the bacterium may also disrupt the blood-brain barrier in mice by dysregulated calcium signaling and gap junctions. This suggests the potential of targeting bacterial crossing of blood-brain barrier for therapeutic use.
The GPCR Grm5 and a downstream factor PLCβ in the calcium signaling pathway show elevation of transcriptional activities in response to infection. Infection of the bacterium Neisseria meningitidis (meningococci) has also been shown to activate calcium signaling (Asmat et al., 2014). It was found that elevation of cytoplasmic calcium concentration elevated in N. meningitides-infected cells is for the adherence of the bacteria to the cells. The activity of PLCβ facilitates the adherence of N. meningitidis through the upregulation of cytoplasmic calcium concentration. This result suggests that B. burgdorferi infection may also harness similar regulatory mechanism used by N. meningitides in elevating cytoplasmic calcium concentration in order to achieve high adherence to blood vessels, facilitating the crossing of the blood-brain barrier for subsequent CNS infection.
Additionally, the activation of Adora2a reveals the dampening of immune response. Adora2a has been shown to be involved in the infection of Plasmodium falciparum, a common pathogen of malaria (Auburn et al., 2010; Gupta et al., 2015). The ligand adenosine activates the Adora2a receptor, which in turn triggers other downstream processes. To prevent cells from over-stimulation, a negative feedback mechanism is launched to impede further excitation by mediating the dissociation of a subunit from Adora2a (Auburn et al., 2010; Metaye et al., 2005). This down-regulation of Adora2a may be to shield the cells from over-excitation. This indicates Adora2a expression could be a biomarker of infection and that anti-inflammatory drugs may exacerbate Lyme treatment.
We also found perturbations in the expression of genes involved in gap junctions. Inflammatory responses due to infection often associate with increased expression of ion channels like gap junctions to facilitate cellular communication (Eugenin et al., 2012). Moreover, studies have shown that gap function regulation plays a role in triggering cell death in virus-infected cells in the CNS (Eugenin et al. 2012). Our results reveal that some neurological responses caused by B. Burgdorferi, including the role of gap junctions, are similar to bacterial and viral infections.
We found differential expression of genes involved in melanogenesis. There is no evidence suggesting melanogenesis and bacterial infection are linked. However, two genes from this pathway, Adcy4 and Plcb1, are also involved with calcium signaling and gap junction pathways. We propose that melanogenesis was highlighted by the two signaling pathway discovery methods because of this overlap.
Enzymes involved in cAMP synthesis, like Adcy4, were perturbed; this likely affects cellular signaling. (Tanaka et al., 2013) conducted a transcriptome analysis of murine brain tissue infected with Toxoplasma gondii, an intracellular pathogenic protozoan. The parasite causes systemic infection and persists in the brain and muscle tissue. They found over 30 genes to be significantly upregulated, including Cxcl9, H2-Eb1, Ccl8, H2-Aa, Zbp1 and Igtp. We observed these genes to be also upregulated in Lyme heart infection. However, no genes were found in both the T. gondii study and on our list of DEGs in the brain. This suggests more work needs to be done to understand the molecular basis of neuroborreliosis.
In conclusion, we present a dual-method pipeline to analyze the host transcriptome Borrelia infection using RNA-seq. Many immune response-related genes were differentially expressed in heart tissue and far fewer were identified in the brain. We propose that Borrelia may disrupt the blood-brain barrier in mice and induces a peripheral inflammatory cascade.
First, although infection was not established in the brain, the tissue is affected as many genes are differentially expressed and we found that neuronal gap junctions and calcium signaling are disrupted. This is a hallmark of loss of integrity of the blood-brain barrier. Thus, the damage is occurring irrespective of direct brain infection. Moreover, this suggests that in human infection, the crossing of the blood-brain barrier and infection of the central nervous system are two events. It may be possible to study Borrelia’s effect on the blood-brain barrier in mice, even though the central nervous system is not infected in a mouse model of the disease.
Second, none of the predicted cytokine genes were significantly differentially expressed in this experiment even though the chemokine pathway was perturbed by Borrelia infection. This indicates that these cytokines are induced by the peripheral immune response. However, cytokine-cytokine receptor interaction via Gm2023 (Figure 3A) and receptor CD45 were over-expressed in the infected heart tissue (Figure 3B), allowing phagocyte recruitment to destroy infected cells. Thus, the heart tissue is responding to inflammation but is not producing these cytokines. These results not only elucidate the transcriptional basis of self-perpetuating cascade alluded to immunological responses found in Borrelia infection but also affirm the utility of the dual-method approach proposed in our study.
Challenges facing diagnosis and treatment of Lyme are significant. Prolonged symptoms after antibiotic treatment are still afflicting a small percentage of patients, making the topic of “chronic Lyme disease” interesting but understudied. Although the mouse is not a perfect model of human Lyme disease, we show that the mouse can be used to examine unique features of Borrelia infection and the crossing of the blood-brain barrier. A thorough molecular study to explore these pathways over time is needed to elucidate the etiology of lingering Lyme symptoms in the host in order to improve patient outcome.
ACKNOWLEDGEMENTS
We thank the financial support of Lafayette College for MC and EH. Thanks to Drs. Laurie Caslake, Elaine Reynolds, and Robert Kurt for lab facilities, assistance in the infection process, and discussion. Lastly, thank you to Kimberly Olsen of the Baumgarth lab at the University of California, Davis, for providing the Borrelia samples.
REFERENCES
Anders, S., Pyl, P. T., & Huber, W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics, 31(2), 166-169. doi:10.1093/bioinformatics/btu638
Andrews, S. (2010). FastQC A Quality Control tool for High Throughput Sequence Data. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Armstrong, A. L., Barthold, S. W., Persing, D. H., & Beck, D. S. (1992). Carditis in Lyme disease susceptible and resistant strains of laboratory mice infected with Borrelia burgdorferi. Am J Trop Med Hyg 47:249-258.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P, Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., & Sherlock, G. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25-29. doi: 10.1038/75556
Asmat, T. M, Tenenbaum, T., Jonsson, A. B., Schwerk, C., & Schroten, H. (2014). Impact of calcium signaling during infection of Neisseria meningitidis to human brain microvascular endothelial cells. PLoS One 9:e114474. doi: 10.1371/journal.pone.0114474
Auburn, S., Fry, A. E., Clark,T. G., Campino, S., Diakite, M., Green, A., Richardson, A., Jallow, M., Sisay-Joof, F., Pinder, M., Molyneux, M. E., Taylor, T. E., Haldar, K., Rockett, K. A., & Kwiatkowski, D. P. (2010). Further evidence supporting a role for gs signal transduction in severe malaria pathogenesis. PLoS One 5:e10017. doi: 10.1371/journal.pone.0010017
Barthold, S. W., de Souza, M. S., Janotka, J. L., Smith, A. L., & Persing, D. H. (1993). Chronic Lyme borreliosis in the laboratory mouse. Am J Pathol 143:959-971.
Browser UG. Retrived from http://hgdownload.soe.ucsc.edu/goldenPath/mm10/
chromosomes/. CDC. (2014). CDC Statistics. Retrieved from http://www.cdc.gov/lyme/stats/index.html (accessed July 2014).
Centers for Disease C, & Prevention. (1995). Recommendations for test performance and interpretation from the Second National Conference on Serologic Diagnosis of Lyme Disease. MMWR Morb Mortal Wkly Rep 44:590-591.
Chang, B. (2015). Survey of the nob5 mutation in C3H substrains. Mol Vis 21:1101-1105.
Coureuil M, Join-Lambert O, Lecuyer H, Bourdoulous S, Marullo S, and Nassif X. 2013. Pathogenesis of meningococcemia. Cold Spring Harb Perspect Med 3. doi: 10.1101/cshperspect.a012393cummeRbund. Retrieved from http://compbio.mit.edu/cummeRbund/.
Dressler, F., Whalen, J. A., Reinhardt, B. N., & Steere, A. C. (1993). Western blotting in the serodiagnosis of Lyme disease. J Infect Dis 167:392-400.
Eugenin, E. A., Basilio, D., Saez, J. C., Orellana, J. A., Raine, C. S., Bukauskas, F., Bennett, M. V., & Berman, J. W. (2012). The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J Neuroimmune Pharmacol 7:499-518. doi: 10.1007/s11481-012-9352-5
Fraser, C. M., Casjens, S., Huang, W. M., Sutton, G. G., Clayton, R., Lathigra, R., White, O., Ketchum, K. A., Dodson, R., Hickey, E. K., Gwinn, M., Dougherty, B., Tomb, J. F., Fleischmann, R. D., Richardson, D., Peterson, J., Kerlavage, A. R., Quackenbush, J., Salzberg, S., Hanson, M., van Vugt, R., Palmer, N., Adams, M. D., Gocayne, J., Weidman, J., Utterback, T., Watthey, L., McDonald, L., Artiach, P., Bowman, C., Garland, S., Fuji, C., Cotton, M. D., Horst, K., Roberts, K., Hatch, B., Smith, H. O., & Venter, J. C. (1997). Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390:580-586. doi: 10.1038/37551
Garcia-Monco, J. C., Miller, N. S., Backenson, P. B., Anda, P., & Benach, J. L. (1997). A mouse model of Borrelia meningitis after intradermal injection. J Infect Dis 175:1243-1245.
GENEWIZ. (2013). RNA-seq service at South Plainfield, New Jersey, USA. Turnaround time was approximately 6 weeks.
Grab, D. J., Nyarko, E., Nikolskaia, O. V., Kim, Y. V., & Dumler, J. S. (2009). Human brain microvascular endothelial cell traversal by Borrelia burgdorferi requires calcium signaling. Clin Microbiol Infect 15:422-426. doi: 10.1111/j.1469-0691.2009.02869.x
Grab, D. J., Perides, G., Dumler, J. S., Kim, K. J., Park, J., Kim, Y. V., Nikolskaia, O., Choi, K. S., Stins, M. F., & Kim, K. S. (2005). Borrelia burgdorferi, host-derived proteases, and the blood-brain barrier. Infect Immun 73:1014-1022. doi: 10.1128/IAI.73.2.1014-1022.2005
Gupta, H., Jain, A., Saadi, A. V., Vasudevan, T. G., Hande, M. H., D'Souza, S. C., Ghosh, S. K., Umakanth, S., & Satyamoorthy, K. (2015). Categorical complexities of Plasmodium falciparum malaria in individuals is associated with genetic variations in ADORA2A and GRK5 genes. Infect Genet Evol 34:188-199. doi: 10.1016/j.meegid.2015.06.010
Halperin, J. J. (2015). Chronic Lyme disease: misconceptions and challenges for patient management. Infect Drug Resist 8:119-128. doi: 10.2147/IDR.S66739
Huang da, W., Sherman, B. T., & Lempicki, R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1-13. doi: 10.1093/nar/gkn923
Huang da, W., Sherman, B. T., & Lempicki, R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44-57. doi: 10.1038/nprot.2008.211
Jiang, D., Xie, T., Liang, J., & Noble, P. W. (2013). beta-Arrestins in the immune system. Prog Mol Biol Transl Sci 118:359-393. doi: 10.1016/B978-0-12-394440-5.00014-0
Kanehisa, M., & Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27-30.
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., & Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42:D199-205. doi: 10.1093/nar/gkt1076
Langmead, B., & Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357-359. doi: 10.1038/nmeth.1923
Love, M. I., Huber, W., & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8
Luo, W., & Brouwer, C. (2013). Pathview: an R/Bioconductor package for path-way-based data integration and visualization. Bioinformatics 29:1830-1831. doi: 10.1093/bioinformatics/btt285
McAlister, H. F., Klementowicz, P. T., Andrews, C., Fisher, J. D., Feld, M., & Fur-
man, S. (1989). Lyme carditis: an important cause of reversible heart block. Ann Intern Med 110:339-345.
McConville, M. 2014. Open questions: microbes, metabolism and host-pathogen
interactions. BMC biology 12. doi: 10.1186/1741-7007-12-18
Metaye, T., Gibelin, H., Perdrisot, R., & Kraimps, J. L. (2005). Pathophysiologi-
cal roles of G-protein-coupled receptor kinases. Cell Signal 17:917-928. doi: 10.1016/j.cellsig.2005.01.002
Narasimhan, S., Caimano, M. J., Liang, F. T., Santiago, F., Laskowski, M., Philipp,
M. T., Pachner, A. R., Radolf, J. D., & Fikrig, E. (2003). Borrelia burgdorferi transcriptome in the central nervous system of non-human primates. Proc Natl Acad Sci U S A 100:15953-15958. doi: 10.1073/pnas.2432412100
Pachner, A. R., & Steere, A. C. (1984). Neurological findings of Lyme disease. Yale J Biol Med 57:481-483.
Radolf, J. D., Caimano, M. J., Stevenson, B., & Hu, L. T. (2012). Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol 10:87-99. doi: 10.1038/nrmicro2714
Rasigade, J. P., Trouillet-Assant, S., Ferry, T., Diep, B. A., Sapin, A., Lhoste, Y., Ranfaing, J., Badiou, C., Benito, Y., Bes, M., Couzon, F., Tigaud, S., Lina, G., Etienne, J., Vandenesch, F., & Laurent, F. (2013). PSMs of hypervirulent Staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PLoS One 8:e63176. doi: 10.1371/journal.pone.0063176
Rego, R. O., Bestor, A., Stefka, J., & Rosa, P. A. (2014). Population bottlenecks during the infectious cycle of the Lyme disease spirochete Borrelia burgdorferi. PLoS One 9:e101009. doi: 10.1371/journal.pone.0101009
Rosa, P. A., Tilly, K., & Stewart, P. E. (2005). The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol 3:129-143. doi: 10.1038/nrmicro1086
Rupprecht, T. A., Koedel, U., Fingerle, V., & Pfister, H. W. (2008). The pathogenesis of lyme neuroborreliosis: from infection to inflammation. Mol Med 14:205-212. doi: 10.2119/2007-00091.Rupprecht
Soderblom, T., Oxhamre, C., Wai, S. N., Uhlen, P., Aperia, A., Uhlin, B. E., & Richter-Dahlfors, A. (2005). Effects of the Escherichia coli toxin cytolysin A on mucosal immunostimulation via epithelial Ca2+ signalling and Toll-like receptor 4. Cell Microbiol 7:779-788. doi: 10.1111/j.1462-5822.2005.00510.x
Tanaka, S., Nishimura, M., Ihara, F., Yamagishi, J., Suzuki, Y., & Nishikawa, Y. (2013). Transcriptome analysis of mouse brain infected with Toxoplasma gondii. Infect Immun 81:3609-3619. doi: 10.1128/IAI.00439-13
Tarca, A. L., Kathri, P., & Draghici, S. (2013). SPIA: Signaling Pathway Impact Analysis (SPIA) using combined evidence of pathway over-representation and unusual signaling perturbations. R package version 2.20.0.
TranVan Nhieu, G., Clair, C., Grompone, G., & Sansonetti, P. (2004). Calcium signalling during cell interactions with bacterial pathogens. Biol Cell 96:93-101. doi: 10.1016/j.biolcel.2003.10.006
Trapnell, C., Pachter, L., & Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105-1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., Pimentel, H., Salzberg, S. L., Rinn, J. L., & Pachter, L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562-578. doi: 10.1038/nprot.2012.016
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J., & Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511-515. doi: 10.1038/nbt.1621
Wang, G., Petzke, M. M., Iyer, R., Wu, H., & Schwartz, I. (2008). Pattern of proinflammatory cytokine induction in RAW264.7 mouse macrophages is identical for virulent and attenuated Borrelia burgdorferi. J Immunol 180:8306-8315.
Wang, G., van Dam, A. P., Schwartz, I., & Dankert, J. (1999). Molecular typing of Borrelia burgdorferi sensu lato: taxonomic, epidemiological, and clinical implications. Clin Microbiol Rev 12:633-653.
Webb, L. X., Wagner, W., Carroll, D., Tyler, H., Coldren, F., Martin, E., & McSir. (2007). Osteomyelitis and intraosteoblastic Staphylococcus aureus. J Surg Orthop Adv 16:73-78.
WHO. (2006). Lyme borreliosis in Europe. Influences of climate and climate change, epidemiology, ecology and adaptation measures. Retrieved from: http://www.euro.who.int/en/publications/abstracts/lyme-borreliosis-in-